- 产地
- 苏州
- 品牌
- 细胞高效转染试剂
- 型号
- 齐全
- 是否定制
- 是
细胞转染效率低下的几个大坑:1.不注意细节在转染之前更换一次37℃预温的培养基,可提高转染效率。脂质体和DNA/RNA混合物应当一滴一滴地滴下来,从培养皿一边滴到另一边,边滴边轻摇培养皿,以确保均匀分布和避免局部高浓度。这些都是一些细枝末节,但是,如果不注意的话,也会造成转染效率低下。2.细胞状态不好细胞状态不好,会导致转染效率低下。一般进行转染的细胞应该处于对数生长期。如果是贴壁细胞的话,贴壁率应该在70-80%;如果是悬浮细胞的话,应该是6X105个/孔(24孔板),一般是转染前现在换液,转染前用无血清培养基或PBS洗细胞一次。转染的整个过程都不应该有物品。瞬时和稳定表达DNA转染后,转入基因的表达可以在1-4天内检测到。无锡细胞高效转染试剂价格

细胞转染之PEI转染法:1.细胞分盘:通过胰酶消化收集细胞,用适当的完全培养基以1×105至4×105细胞/cm2的密度平铺细胞于35mm培养皿上(根据实验需要选择培养皿,使细胞贴壁后所占面积达到培养皿总面积的70-90%)。将细胞置于含5%CO2的37℃温箱中孵育8-24h,当细胞贴壁完全后即可开始转染。转染前2h换液(用1.5ml无血清培养基置换旧的培养基)。注:PEI转染法对细胞生长有克制作用,在换液前细胞几乎不生长,所以需要密度比较大时做转染。2.制备PEI-DNA混合物以35mm组织培养皿用240μl反应总体积为例。先在无菌EP管中加入120μl1×HBS,再加入质粒DNA(总量1-3μg为佳),混匀后加入等体积PEI工作液,充分混匀后室温静置20-30min,较后将这240μl的PEI-DNA混合液逐滴加入上述单层细胞的细胞培养基中,轻轻摇动平皿混匀后置于含5%CO2的37℃温箱孵育。厦门细胞高效转染试剂厂家转染试剂的准备①将400ul去核酸酶水加入管中,震荡10秒钟,溶解脂状物。

细胞转染效率低下的几个大坑:1.试剂过期有时候转染效率低下,还要考虑会不会存在试剂过期的情况。毛博当年做转染整整半年,百思不得其解。较后发现,原来是Lipofectamine2000过期了。换了之后,立马成功。根据我的经验,尽量使用开封半年内的,因为过了半年后,就算没有过失效期,但是细胞毒性较大增加,而转染效率却较大下降。千万不要为了省钱,影响实验进度哈。2.未加血清转染后未及时加入血清,会导致细胞大量死亡。一般要在转染后的4-6小时换液且换为有血清的培养基。根据毛博的经验,其实也可以在原来的无血清培养基里面滴加血清。这个时候,较好不要换液,不要打扰细胞,让它安安静静地休息。但是也不能过早加入血清
如何高效率实现细胞转染:DNA转染导入细胞胞浆内的DNA不能穿过细胞核的核膜("核屏障")。在细胞有丝分裂过程中核膜溶解,DNA进入细胞核才有可能。为此,细胞处于分裂期对DNA转染是至关重要的,并且处于分裂期的细胞比例必须尽可能大才能实现高效转染。DNA转染机制示意图如下所示:转染RNA(siRNA/miRNA/mRNA)对于RNA转染是没有"核屏障"的,因为RNA不需要进入细胞核发挥其生物学效应,因此细胞分裂对它没有影响。转染试剂的选择理想的转染试剂选择可以使您的实验如虎添翼!真核细胞的先天免疫系统,使它们能够检测外来物质如脂多糖、细菌或细菌核酸和蛋白质,并克制潜在病原体的入侵。此外,细胞通过信使分子向临近细胞传递发现有害物质的信号。因此,这些临近细胞甚至无需接触病原体即可采取防御方式。这种细胞先天免疫系统也是转染成功的一个障碍。大部分外源DNA会被核酸酶降解或随细胞分裂而稀释。

细胞转染效率影响因素:1.细胞密度一般来说,当细胞密度达到60%~80%时进行转染可以取得较高的转染效率,过低或过高都会影响转染效果。2.细胞生长状态这点非常关键,很多时候传代次数太少或者太多都将导致细胞对转染试剂敏感度的改变。因此,为了提高转染效率以及转染稳定性、降低细胞毒性,应尽量使用适度传代的细胞系,并在不同次实验时保持细胞传代次数的一致性。3.细胞种类不同细胞种类的细胞在做转染时所需要的转染体系是不同的,不能一种体系用在所有类型细胞的转染实验。瞬时转染后,可在48h-72h细胞长满之后,提取细胞蛋白或者RNA。南昌正规细胞高效转染试剂厂家供应
代细胞和传代次数多的细胞都不是较佳选择。无锡细胞高效转染试剂价格
转染的注意事项:培养基中的物品物品,比如青霉素和链霉素,是影响转染的培养基添加物。这些物品一般对于真核细胞无毒,但阳离子脂质体试剂增加了细胞的通透性,使物品可以进入细胞。这降低了细胞的活性,导致转染效率低。所以,在转染培养基中不能使用物品,甚至在准备转染前进行细胞铺板时也要避免使用物品。这样,在转染前也不必润洗细胞。对于稳定转染,不要在选择性培养基中使用青霉素和链霉素,因为这些物品是GENETICIN选择性物品的竞争性克制剂。无锡细胞高效转染试剂价格

细胞转染常用步骤:1.转染试剂的准备①将400ul去核酸酶水加入管中,震荡10秒钟,溶解脂状物。②震荡后将试剂放在-20摄氏度保存,使用前还需震荡。2.选择合适的混合比例(1:1-1:2/脂质体体积:DNA质量)来转染细胞。在一个转染管中加入合适体积的无血清培养基。加入合适质量的MyoD或者EGFP的DNA,震荡后在加入合适体积的转染试剂,再次震荡。3.将混合液在室温放置10―15分钟。4.吸去培养板中的培养基,用PBS或者无血清培养基清洗一次。5.加入混合液,将细胞放回培养箱中培养一个小时。6.到时后,根据细胞种类决定是否移除混合液,之后加入完全培养基继续培养24-48小时使用基质珠做为固相...
- 开封正规细胞高效转染试剂服务电话 2026-01-02
- 宁波正规细胞高效转染试剂报价 2026-01-02
- 深圳正规细胞高效转染试剂直销价 2026-01-01
- 深圳细胞高效转染试剂哪家好 2026-01-01
- 青岛细胞高效转染试剂推荐厂家 2026-01-01
- 厦门唐山细胞高效转染试剂 2026-01-01
- 广州细胞高效转染试剂厂家批发价 2026-01-01
- 武汉细胞高效转染试剂价格 2025-12-31
- 南京广州细胞高效转染试剂 2025-12-31
- 贵阳细胞高效转染试剂厂家推荐 2025-12-31


- 北京细胞高效转染试剂价格 2025-12-31
- 宁波正规细胞高效转染试剂进货价 2025-12-31
- 济南正规细胞高效转染试剂哪里买 2025-12-31
- 厦门正规细胞高效转染试剂厂家推荐 2025-12-31
- 苏州细胞高效转染试剂生产厂家 2025-12-31
- 唐山正规细胞高效转染试剂直销厂家 2025-12-31



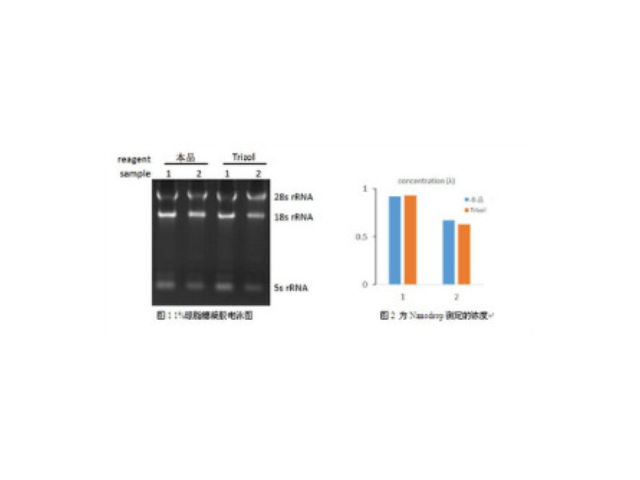
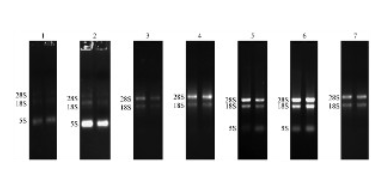
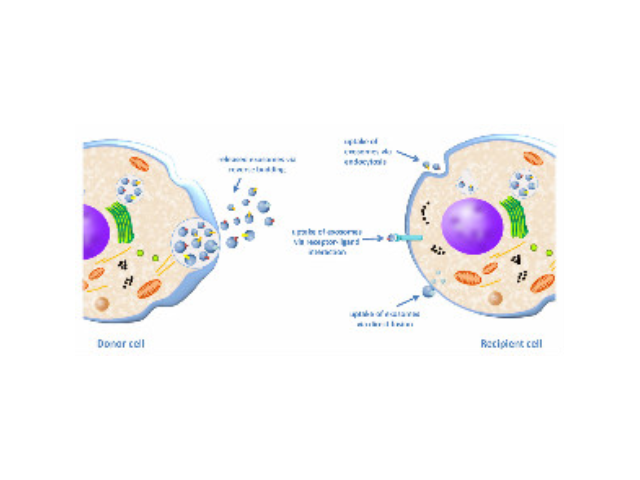
- 天津正规细胞外基质胶哪家便宜 01-05
- 唐山正规外泌体提取试剂哪里买 01-05
- 济南RNA提取试剂哪家便宜 01-05
- 郑州外泌体提取试剂价格 01-05
- 杭州正规鼠尾胶原进货价 01-05
- 宁波外泌体提取试剂直销价 01-05
- 苏州正规RNA提取试剂哪家好 01-05
- 上海正规糖原染色试剂盒直销价 01-05
- 温州外泌体提取试剂销售厂家 01-05
- 南京贵阳细胞外基质胶 01-05