- 产地
- 苏州
- 品牌
- 细胞高效转染试剂
- 型号
- 齐全
- 是否定制
- 是
如何有效提高细胞转染实验效率:1.选择高效的转染方法不同转染试剂有不同的转染方法,但大多大同小异。转染时应跟据具体转染试剂推荐的方法,但也要注意,因不同实验室培养的细胞性质不同,质粒定量差异,操作手法上的差异等,其转染效果可能不同,应根据实验室的具体条件来确定较佳转染条件。2.确保所构建载体的质量。转染载体的构建(细菌载体,质粒DNA,RNA,PCR产物,寡核苷酸等)也影响转染结果。细菌载体对特定宿主细胞传染效率较高,但不同细菌载体有其特定的宿主,有的还要求特定的细胞周期,如逆转录细菌需侵染分裂期的宿主细胞,此外还需考虑一些安全问题(如基因污染)。除载体构建外,载体的形态及大小对转染效率也有不同的影响,如前面提到的超螺旋及线性DNA对瞬时和稳定转染的影响。如果基因产物对细胞有毒性作用,转染也很难进行,因此选择组成或可调控,强度合适的启动子也很重要,同时做空载体及其它基因的相同载体构建的转染正对照可排除毒性影响的干扰。其可降解性对体内应用也具有重要的意义。宁波正规细胞高效转染试剂哪家好

细胞转染的常用方法:细菌转染:原理:对于用脂质体不能实现转染的细胞,可以采用细菌转染。可用于蛋白质过表达或克制,是临床研究中较常用的方法。它通过侵染宿主细胞将外源基因整合到染色体中,可用于难转染细胞、原代细胞的稳定性转染。实验步骤:通过基因克隆生成重组细菌 → 采用非细菌法转染包装细胞系,扩增并分离得到重组细菌颗粒→纯化并滴定细菌液→转导目的细胞(含有细菌特异性的受体)→去除培养物中的细菌,并加入新鲜培养基→分析细胞瞬时基因表达或沉默情况。无锡正规细胞高效转染试剂厂家现货果观察到转染效率降低,可以试着转染新鲜培养的细胞以恢复较佳结果。

细胞转染实验简介:实验原理外源基因进入细胞主要有四种方法:电击法、磷酸钙法和脂质体介导法和细菌介导法。电击法是在细胞上短时间暂时性的穿孔让外源质粒进入;磷酸钙法和脂质体法是利用不同的载体物质携带质粒通过直接穿膜或者膜融合的方法使得外源基因进入细胞;细菌法是利用包装了外源基因的细菌传染细胞的方法使得其进入细胞。但是由于电击法和磷酸钙法的实验条件控制较严、难度较大;细菌法的前期准备较复杂、而且可能对于细胞有较大影响;所以现在对于很多普通细胞系,一般的瞬时转染 方法多采用脂质体法。
DNA细胞转染步骤:1.提前现在细胞铺板 提前现在将细胞种植在24孔板中,以转染时细胞密度在60%左右为宜。2.转染过程⑴将0.8μg的DNA用25μl无血清稀释液稀释,充分混匀,制成DNA稀释液。 注意:无血清稀释液建议采用OPTI-MEM、无血清DMEM或1640。⑵将2μlEntransterTM-H4000用25μl无血清稀释液体稀释,充分混匀,制成EntransterTM-H4000稀释液。室温静置5分钟。⑶将EntransterTM-H4000稀释液分别加入到DNA稀释液中,充分混匀(可用振荡器振荡或用加样器吹吸10次以上),室温静置15分钟。转染复合物制备完成。⑷将转染复合物加入到含细胞和完全培养基的培养容器上,轻柔混匀。 注意:①对本试剂,采用含血清的全培养基有助于提升转染效率。②完全培养基可加物品。可以使用一些营养丰富的无血清培养基,或者在转染培养基中使用血清。

细胞转染效率影响因素:1.细胞密度一般来说,当细胞密度达到60%~80%时进行转染可以取得较高的转染效率,过低或过高都会影响转染效果。2.细胞生长状态这点非常关键,很多时候传代次数太少或者太多都将导致细胞对转染试剂敏感度的改变。因此,为了提高转染效率以及转染稳定性、降低细胞毒性,应尽量使用适度传代的细胞系,并在不同次实验时保持细胞传代次数的一致性。3.细胞种类不同细胞种类的细胞在做转染时所需要的转染体系是不同的,不能一种体系用在所有类型细胞的转染实验。转染试剂与细胞不匹配细胞转染较适合的不是原代细胞。金华正规细胞高效转染试剂厂家现货
转化、转染、转导几个名词是从事生命科学研究的初学者经常碰到的。宁波正规细胞高效转染试剂哪家好
细胞转染效率低下的几个大坑:1.准备不足做脂质体转染的时候,在开展正式实验前要多做预试验,优化转染条件。优化转染条件包括:脂质体的用量、DNA密度、细胞密度、脂质体和DNA混合孵育时间等等。2.电压过大做电转的时候,如果电压太大,往往会发生细胞大量死亡的情况。不同的细胞,需要的电压是不一样的。这就要求我们多做预实验,多摸索条件。对于大多数细胞来说,其较佳电压位于250-1250v/cm。另外,就是进行转染的细胞应该处于对数生长期。处于对数生长期的细胞的抗损伤能力是较强的。细胞浓度应该处于5x106到1x107/mL之间。每次转染的质粒应该控制在4-6 μg,如果﹥10μg,转染效率也较大降低。电击后,应该将DNA和细胞混合液在室温下放置10分钟,使细胞恢复损伤。宁波正规细胞高效转染试剂哪家好

细胞转染的注意事项:细胞状态这点非常重要,不要急于求成,一定要让细胞处于较佳的生长状态再做。有文献说传代不要超过17代。细胞复苏后的3代左右时细胞状态较好,不要用传了很多代的细胞去做,细胞的形态都会发生变化。大多数已建立的细胞系都是非整倍体,原代培养包括了表达不同基因组合的细胞的混合物。细胞培养在实验室中保存数月和数年后会经历突变,总染色体重组或基因调控变化等而演化。这会导致和转染相关的细胞行为的变化。如果随时间发现这种变化,融化一管新鲜的细胞可能会恢复原先的转染活性。因此,如果观察到转染效率降低,可以试着转染新鲜培养的细胞以恢复较佳结果。或者,几种来源于经筛选,转染效率较高细胞亚系的细胞系现...
- 珠海细胞高效转染试剂价格 2026-04-30
- 苏州正规细胞高效转染试剂服务电话 2026-04-30
- 细胞高效转染试剂推荐厂家 2026-04-30
- 长沙细胞高效转染试剂直销价 2026-04-29
- 重庆正规细胞高效转染试剂推荐厂家 2026-04-28
- 重庆正规细胞高效转染试剂哪家便宜 2026-04-27
- 无锡正规细胞高效转染试剂产品介绍 2026-04-26
- 细胞高效转染试剂销售厂家 2026-04-26
- 徐州正规细胞高效转染试剂进货价 2026-04-25
- 苏州细胞高效转染试剂厂家批发价 2026-04-25


- 广州细胞高效转染试剂报价 2026-04-23
- 北京正规细胞高效转染试剂销售厂家 2026-04-23
- 南昌细胞高效转染试剂供应商 2026-04-23
- 石家庄细胞高效转染试剂单价 2026-04-23
- 南京细胞高效转染试剂单价 2026-04-23
- 宁波石家庄细胞高效转染试剂 2026-04-23


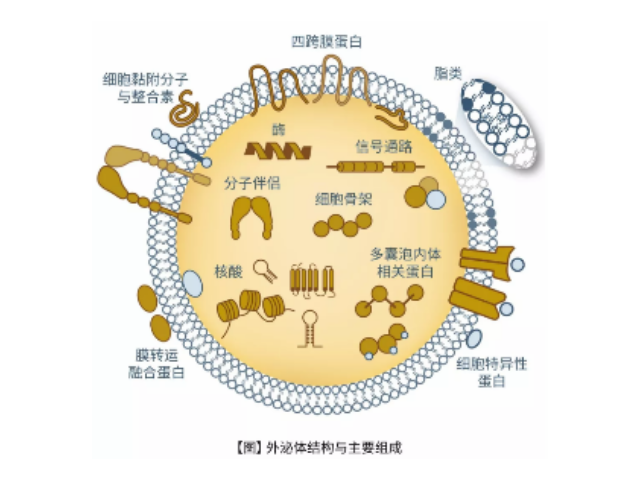



- 石家庄原代细胞分离试剂盒厂家现货 05-01
- 成都原代细胞分离试剂盒直销价 05-01
- 广州正规原代细胞分离试剂盒价格 05-01
- 杭州原代细胞分离试剂盒销售厂家 05-01
- 开封正规原代细胞分离试剂盒厂家批发价 05-01
- 珠海原代细胞分离试剂盒直销厂家 05-01
- 芜湖郑州细胞高效转染试剂 05-01
- 深圳正规原代细胞分离试剂盒哪家便宜 05-01
- 广州原代细胞分离试剂盒平均价格 05-01
- 贵阳外泌体提取试剂厂家批发价 05-01