内毒素检测结果误差可能源于多环节:试剂方面,鲎试剂(LAL )或试剂批间差异、过期试剂活性下降会导致结果偏差,需通过试剂验收(如阳性对照回收率验证)确保质量;操作方面,实验器具未除热原(如玻璃器皿未干热灭菌)、加样体积不准确会引入污染或误差,需严格执行 SOP(如器皿 250℃干热灭菌≥30 分钟);环境方面,实验室空气中的微生物孢子、粉尘可能污染样品,需在洁净工作台操作并设置阴性对照。此外,反应温度波动(偏离 37℃±1℃)会影响酶活性,需使用恒温孵育器精确控温,确保反应条件稳定。
湖州申科内毒素检测服务含低内毒素回收,通过不同样品前处理方式搭配多种检测方法,较大程度解决LER问题。化学制药内毒素检测法规要求
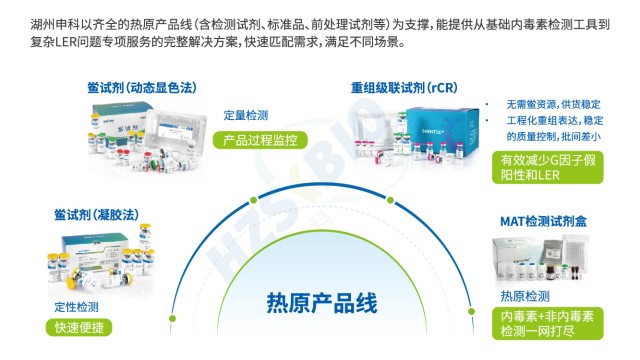
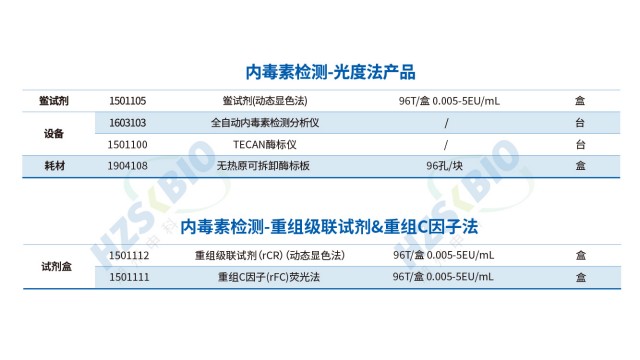
在內毒素检测的技术体系中,凝胶法与动态显色法基于不同原理与特性,形成互补应用格局。凝胶法依托鲎试剂与内毒素的凝集反应,实现定性或半定量检测,其灵敏度覆盖 0.03EU/ml、0.06EU/ml 等多梯度,60 分钟即可完成反应;检测结果依赖肉眼观察(180° 倒转判读凝胶形成),数据需手工记录,配套 内毒素凝胶法测定仪(恒温仪) 即可开展,虽自动化程度有限,但操作简洁,适用于生产环节的快速初筛。与之相比,动态显色法通过监测反应混合物吸光度或透光率的变化(如达预设检测值的反应时间、信号增速)实现 定量检测 ,灵敏度拓展至 5-0.005EU/ml ,60-90 分钟反应时长虽略长,却可借助酶标仪或全自动内毒素检测分析仪完成全流程自动化操作—软件实时采集数据,契合药品生产质量管理规范(GMP)对数据追溯与精度的要求。二者各有侧重:凝胶法以 “快速定性” 服务基础防控,动态显色法凭 “准确定量 + 自动化” 支撑严苛质控,共同为內毒素检测提供灵活适配的技术路径。
浙江原料药内毒素检测法规要求内毒素检测需关注制剂成分,螯合剂和表面活性剂可能诱发“低内毒素回收(LER)”。
在为新物料或新产品、中间产品建立细菌内毒素检测方法时,常会遇到各种困难,尤其是尚处于新药研发早期阶段的药物。此时,由于药物制剂、缓冲系统等还不稳定,经常会发生变化,这样就给方法的建立带来了不同程度的影响。在建立细菌内毒素检查法之前,须尽可能多地了解有关该药品的基本信息,例如:有关样品的可溶性信息、推荐的稀释液、在水中的溶解度以及合适溶剂,样品的pH范围,分子量大小;如果是蛋白产品,还要了解该产品的等电点,产品规格、体积或重量,拟用于临床的用法和用量等,以便选择合适的样品处理方法和内毒素检测方法。
β- 葡聚糖是鲎试剂(LAL)检测内毒素的常见干扰物,可活化 LAL 中的 G 因子通路,导致假阳性结果。干扰多见于含植物源原料的样品(如中药注射剂)、生物发酵产物或环境真菌污染的样品。消除方法包括:使用特异性 LAL 试剂(如添加葡聚糖抑制剂的 LAL),其只对内毒素敏感而不受 β- 葡聚糖影响;采用加热处理(如 80℃加热 10 分钟)破坏 β- 葡聚糖结构;或通过亲和层析去除样品中的 β- 葡聚糖。检测时需设置 β- 葡聚糖阳性对照,若对照反应阳性而内毒素标准品无反应,表明存在干扰,需优化前处理步骤后重新检测。
外源性热原含细菌内毒素、脂磷壁酸、酵母多糖等,单核细胞活化反应检查法可检出全部热原。
检测细菌内毒素的堂试剂方法,是一个生物反应过程,受到很多因素的干扰。在一个供试品的检测方法固定下来之前,为了得到准确的结果,必须要了解供试品与鲎试剂之间的相互关系。供试品中的成分往往非常复杂,而且会干扰试验检测系统的功能。很多干扰的机理,并不是非常清楚。但是业界比较能够接受的理论是,如果供试品中某些因子影响了鲎试剂中蛋白的表达功能,则被认为是干扰作用(Inhibition/Enhancement,V/E)。干扰作用产生的因素较多,一般包括试剂因素(鲎试剂、内毒素标准品)供试品因素(pH值、温度、离子强度、浓度、水溶性、黏度、可发生鲎试剂反应的非内毒素杂质)和实验因素(试验器皿、细菌内毒素检查用水、鲎试剂抗干扰能力)等。
内毒素检查用水经二次精制,无菌无热原,避免检测假阳假阴。合规性内毒素检测结果判定
含蛋白酶的样本致假阳性时,可稀释后 70℃加热 5-15min 灭活,再做内毒素检测。化学制药内毒素检测法规要求
内毒素检测方法验证需覆盖多项参数,确保方法可靠:线性范围需包含样品预期浓度(如 0.01-10 EU/mL),相关系数 R²≥0.98;准确度通过加标回收率评估,应在 50%-200% 范围内;精密度包括批内和批间精密度,CV 值均应≤15%;检测限(LOD)需低于产品限值的 1/2(如限值 0.5 EU/mL,LOD 应≤0.25 EU/mL);专属性需证明无干扰物质影响(如 β- 葡聚糖、蛋白质不引发假阳性)。验证通过后,方法需经实验室负责人批准方可使用,且定期需进行一次回顾性验证,确认方法持续有效。
化学制药内毒素检测法规要求