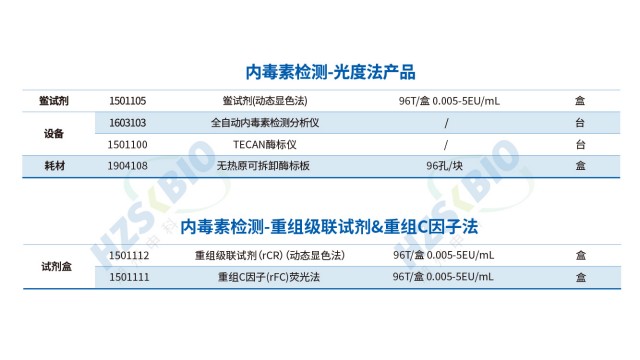
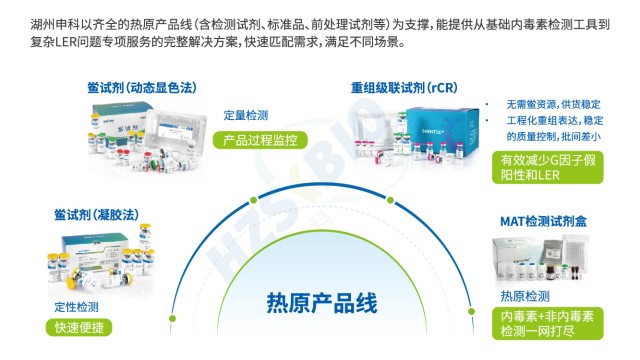
SHENTEK®重组级联试剂为内毒素检测高效解决方案。法规层面,符合美国药典(USP)、欧洲药典(EP)及日本药典(JP)要求,满足国际标准检测需求。抗干扰性强,与天然鲎具有相同反应机制,检测结果等效,适配复杂样本场景。特异性表现突出,不含 G 因子,避免与 β - D 葡聚糖反应产生假阳性,保障结果可靠。检测灵敏度高,线性范围达 0.005 - 5EU/mL ,可准确捕捉低浓度内毒素。反应快速,60分钟内即可完成检测,提升检测效率。兼容性佳,能兼容动态显色法的酶标仪,无需额外投入设备成本。关键的是,无动物源试剂,遵循 3R 原则(减少、替代、优化),不依赖鲎血,解决资源依赖问题,实现长期稳定供应。且通过重组表达生产,批次差异小,重复性好,稳定性高,为生物制品、制药等行业内毒素检测提供可靠、可持续的工具,助力企业把控质量,契合绿色环保与高效检测的双重需求,在保障检测准确性与合规性的同时,推动行业向更环保、更高效方向发展。
内毒素检测方法学验证需覆盖线性、精密度,确保不同批次检测结果稳定。非动物源内毒素检测合规申报
内毒素检测重组级联试剂(rCR)与天然鲎试剂在原料、性能和可持续性上存在本质区别。原料方面,天然鲎试剂依赖鲎血采集,受动物资源限制,而 rCR 通过基因工程表达 C、B 因子及凝固酶原,无动物源性,供应稳定。特异性上,天然鲎试剂因含 G 因子,易与 β-D 葡聚糖反应产生假阳性,而 rCR 剔除 G 因子,只对内毒素特异性响应,从机制上消除干扰。批间一致性方面,天然鲎试剂受鲎个体差异影响,批间 CV 值较高;rCR 成分明确且生产工艺标准化,批间差异明显降低,CV 值≤15%。两者灵敏度相当(0.005EU/mL),但 rCR 无需面临鲎资源政策限制,更符合动物福利趋势和长期质控需求,是天然鲎试剂的理想替代。
非动物源内毒素检测风险评估进行重组级联试剂时,不同酶标仪检测样本 Onset time 有差异,因信号采集方式和灵敏度不同。
内毒素检测的实验方案需遵循“标曲可靠性验证(灵敏度复核)-稀释倍数计算-干扰试验”的完整流程,以保障检测结果准确合规。首先进行标曲可靠性试验,用标准内毒素制备至少3个浓度的稀释液(相邻浓度间稀释倍数不超过10,下限浓度不低于鲎试剂标示检测限),每浓度设 3 支平行管,同时做2支阴性对照;当阴性对照的吸光度小于或透光率大于标准曲线下限的检测值或反应时间大于标准曲线下限的反应时间,对数据进行线性回归,相关系数r的幅值≥0.980时试验方有效,否则需重新操作。接着按公式 L=K/M 确定样本内毒素限值,K 值依给药途径而定,M结合人均60kg体重、1.62m² 体表面积及上限给药剂量计算,也可参考药典行标。随后明确有效稀释上限倍数(MVD),即供试品允许稀释的上限倍数,确保在此范围内检测限值。再开展样本干扰试验,制备供试品溶液(A)、供试品加标溶液(B,加标浓度为标曲中点)、标准曲线溶液(C)及阴性对照(D),按公式 R=(Cs-Ct)/λm×100%计算加标回收率,50%-200%为合格;若鲎试剂、生产工艺等发生变化,需重新进行干扰试验,保障内毒素检测的可靠性。
低内毒素回收(LER)又称内毒素掩蔽,是指无菌制剂(尤其蛋白类生物制剂)进行内毒素检测时,加标内毒素的回收率<50% 的现象,且无法通过稀释排除,区别于传统检测干扰。LER 会导致内毒素污染被低估,已被全球监管机构重点关注:FDA 2013 年要求生物药 BLA 申报时提交 LER 研究报告;EMA 2023 年明确含表面活性剂(如吐温)或螯合剂(如 EDTA)的制剂需提供 LER 数据;中国药典 2025 版 9251 通则也新增 LER 内容,与国际接轨。这些要求促使企业优化内毒素检测流程,避免因 LER 导致的检测偏差,确保药品安全。
细菌内毒素工作标准品可用于鲎试剂灵敏度复核、干扰试验,适配凝胶法与光度法实验。
内毒素检测法规体系正逐步向非动物源试剂倾斜,为重组试剂的应用铺平道路。美国药典(USP)已将重组 C 因子(rFC)和重组级联试剂(rCR)正式收录,于2025 年 5 月纳入 USP-NF,明确要求用户验证重组试剂对特定产品的适用性。欧洲药典(EP)通则 2.6.32 已收录重组 C 因子法,规定注射用水和纯化水可直接采用该方法检测,复杂基质样品需通过验证后使用。日本药原则通过指导原则<G4-4-180>将重组蛋白试剂列为内毒素检测的补充方法。中国药典虽暂未正式收录重组方法,但 9251《细菌内毒素法应用指导原则》为其应用提供了框架。这些法规进展共同构建了重组试剂的合规应用基础。
开展细菌内毒素检测的操作过程,需防止样品受到外源性污染。化学制药内毒素检测方法验证
细菌内毒素是革兰氏阴性菌细胞壁脂多糖,细菌死亡裂解释放,微量级即致人体发热、休克等威胁。非动物源内毒素检测合规申报
当实验室更换内毒素检测方法或更换试剂供应商时,需进行方法比对与桥接验证。比对实验需选取至少 3批代表性样品,分别用新旧方法检测,计算结果相关性(如相关系数 R²≥0.95)和偏差(≤20%)。桥接验证还需评估新方法的特异性、灵敏度是否与旧方法一致,如确认对高风险样品(如含 β- 葡聚糖的样品)的抗干扰能力相当。若方法变更涉及法规申报产品,需将验证数据纳入申报资料,证明变更后方法仍能有效控制内毒素风险,符合 FDA、NMPA 等监管机构对方法变更的合规性要求。
非动物源内毒素检测合规申报