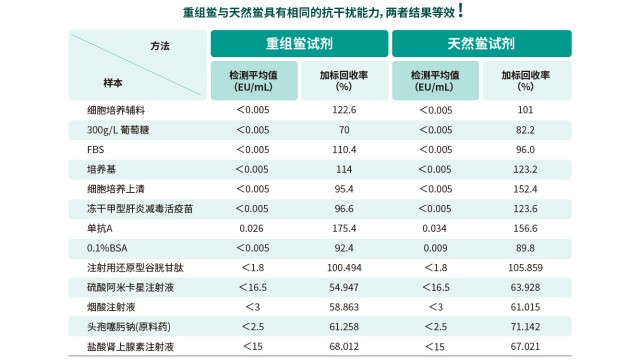
样品中的高渗透性成分(如高浓度盐、糖)会通过改变反应体系渗透压,抑制鲎试剂反应,影响内毒素检测结果。例如,浓度为 70% 的葡萄糖溶液、高浓度氯化钠溶液等,会形成高渗透压环境,导致鲎试剂中的蛋白质脱水变性,丧失酶活性,进而使内毒素无法被正常检测,出现假阴性。这类高渗透性基质的干扰机制明确 —— 通过破坏蛋白质结构影响酶促反应,且干扰程度与浓度正相关。为消除这类干扰,解决方案是使用内毒素检查用水稀释样品:根据样品渗透性高低,逐步稀释至适宜浓度(通常需稀释至渗透压与生理盐水接近),降低对蛋白质的脱水作用,恢复鲎试剂中酶的活性。稀释过程中需注意,稀释倍数需在方法验证确定的 “无干扰稀释范围” 内,避免因过度稀释导致内毒素浓度低于检测限,确保内毒素检测既能规避高渗透性抑制,又能准确捕捉微量内毒素。
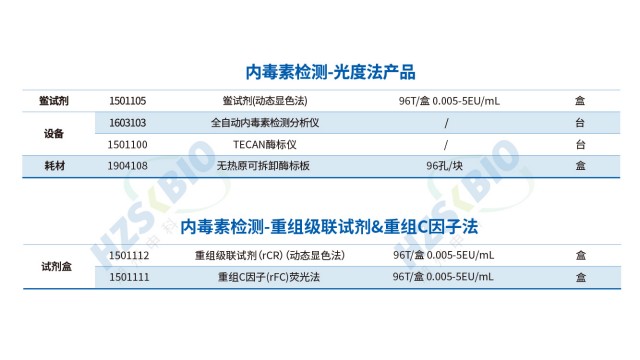
细菌内毒素检查有凝胶法和光度测定法,结果有争议时,除规定外以凝胶限度试验为准。浙江疫苗内毒素检测合规申报
细菌内毒素是革兰氏阴性菌细胞壁外膜释放的脂多糖(LPS)成分,具有极强的生物活性,微量即可引发人体发热、休克甚至多脏器功能衰竭。因此,在生物制品、医疗器械、制药用水等领域,细菌内毒素检测是保障产品安全性的关键质控环节。其检测原理基于内毒素与特定试剂的特异性反应:内毒素可活化鲎血变形细胞裂解物(LAL)中的凝血级联反应,通过C因子通路触发酶促反应,通过观察凝胶形成、浊度变化或显色强度实现定量或定性分析。目前,各国药典均将内毒素检测列为强制要求,以降低临床应用中的热原风险。
浙江疫苗内毒素检测合规申报重组级联试剂(rCR)无动物源,不含 G 因子,可避免 β- 葡聚糖致内毒素检测假阳性。
内毒素检测方法易受样品基质干扰,法规要求在方法应用前必须进行干扰验证,选择标准曲线中点或一个靠近中点的内毒素浓度,作为供试品干扰试验种添加的内毒素浓度计算回收率,若回收率在 50%-200% 范围内,表明无明显干扰;若回收率异常,需通过过滤、中和、透析、加热处理等方式优化样品前处理。方法学确认还需涵盖线性范围(如 0.005-5 EU/mL)、精密度(批内 CV≤15%)、检测限(LOD≤0.01 EU/mL)等指标,确保方法在实际样品检测中稳定可靠,符合《美国药典》 <85>、《中国药典》通则 1143 等药典要求。
生物制品(如单抗、疫苗、重组蛋白)注射剂因直接进入人体,对细菌内毒素残留限值要求严苛(通常≤0.5 EU/mg 或更低),检测面临基质复杂、干扰物质多等挑战。样品中常见的蛋白质、螯合剂、表面活性剂等可能抑制或增强 LAL 反应,需通过预处理消除干扰:如采用稀释法降低基质浓度、添加中和剂(如 Mg²⁺)修复反应体系,或使用热灭活去除蛋白类干扰物。此外,生物制品生产全流程需进行内毒素监控,从细胞培养上清、纯化中间品到终产品均需检测,确保工艺去除内毒素的有效性,符合 ICH Q6B 等法规对 “关键质量属性” 的控制要求。
“低内毒素回收(LER)”可能导致内毒素检测假阴性,对患者用药安全构成潜在隐患。
鲎试验法(LAL 法)是细菌内毒素检测的经典方法,依据反应监测方式可分为三类:凝胶法、浊度法和显色法。凝胶法通过观察是否形成凝胶判断内毒素是否超标,其优势是操作简便、成本较低,适用于定性或半定量检测,如医疗器械初筛;浊度法通过监测反应体系浊度变化速率定量内毒素,灵敏度高(可达 0.005 EU/mL),适用于生物制品原液等高精度需求场景;显色法基于显色底物的吸光度变化定量,抗干扰能力较强,适配复杂基质样品(如含蛋白质的注射液)。三种方法均需严格控制反应温度(37℃±1℃)和时间,确保结果可靠性。
塑料器皿对内毒素吸附力强,制备内毒素工作标准品(CSE)稀释液建议用硼硅酸盐玻璃管。浙江疫苗内毒素检测合规申报
细菌内毒素检查用水需符合灭菌注射用水标准,内毒素含量有明确限值且无干扰。浙江疫苗内毒素检测合规申报
当实验室更换内毒素检测方法或更换试剂供应商时,需进行方法比对与桥接验证。比对实验需选取至少 3批代表性样品,分别用新旧方法检测,计算结果相关性(如相关系数 R²≥0.95)和偏差(≤20%)。桥接验证还需评估新方法的特异性、灵敏度是否与旧方法一致,如确认对高风险样品(如含 β- 葡聚糖的样品)的抗干扰能力相当。若方法变更涉及法规申报产品,需将验证数据纳入申报资料,证明变更后方法仍能有效控制内毒素风险,符合 FDA、NMPA 等监管机构对方法变更的合规性要求。
浙江疫苗内毒素检测合规申报