医疗器械(如输液器、注射器、植入式设备)若携带内毒素,可能通过血液、组织接触引发异常反应或炎症反应。其检测需遵循 “模拟临床使用” 原则:采用浸提液(如 0.9% 氯化钠溶液或注射用水)在 37℃±1℃下浸提器械表面内毒素,再通过 LAL 或 rFC 法检测浸提液。不同器械的内毒素限值差异明显:一次性输液器需≤0.5 EU/device,植入式心脏瓣膜则要求更严格(≤0.06 EU/device)。检测时需注意器械材质对浸提效率的影响,如塑料类器械可能吸附内毒素,需优化浸提时间(通常≥1 小时)或采用超声辅助提取,确保残留内毒素被充分检出。
内毒素检测的方法多样性、多影响因素及实验干扰,会导致自检数据与厂家数据存在差异。北京合规性内毒素检测低内毒素回收
内毒素检测的实验方案需遵循“标曲可靠性验证(灵敏度复核)-稀释倍数计算-干扰试验”的完整流程,以保障检测结果准确合规。首先进行标曲可靠性试验,用标准内毒素制备至少3个浓度的稀释液(相邻浓度间稀释倍数不超过10,下限浓度不低于鲎试剂标示检测限),每浓度设 3 支平行管,同时做2支阴性对照;当阴性对照的吸光度小于或透光率大于标准曲线下限的检测值或反应时间大于标准曲线下限的反应时间,对数据进行线性回归,相关系数r的幅值≥0.980时试验方有效,否则需重新操作。接着按公式 L=K/M 确定样本内毒素限值,K 值依给药途径而定,M结合人均60kg体重、1.62m² 体表面积及上限给药剂量计算,也可参考药典行标。随后明确有效稀释上限倍数(MVD),即供试品允许稀释的上限倍数,确保在此范围内检测限值。再开展样本干扰试验,制备供试品溶液(A)、供试品加标溶液(B,加标浓度为标曲中点)、标准曲线溶液(C)及阴性对照(D),按公式 R=(Cs-Ct)/λm×100%计算加标回收率,50%-200%为合格;若鲎试剂、生产工艺等发生变化,需重新进行干扰试验,保障内毒素检测的可靠性。
上海疫苗内毒素检测技术服务湖州申科内毒素检测服务含低内毒素回收,通过不同样品前处理方式搭配多种检测方法,较大程度解决LER问题。
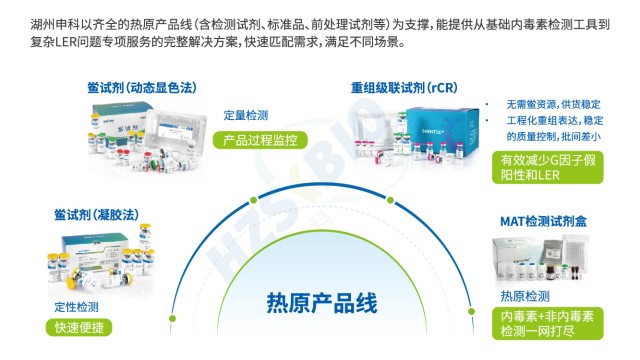
随着生物制药行业的快速发展,注射用药剂、疫苗等制剂及植入性医疗器械的生产需求激增,直接推动了内毒素检测试剂的市场需求。然而,传统内毒素检测严重依赖天然鲎试剂,而全球鲎资源正面临衰减危机。2021 年 2 月,我国国家林业和草原局、农业农村部联合公告将鲎科动物列为国家二级保护动物,严格限制捕捞配额,导致天然鲎试剂价格持续上涨,甚至出现关键检测环节的供应短缺问题。在此背景下,湖州申科研发的重组级联试剂(rCR)通过基因工程技术实现无动物源性生产,彻底摆脱对鲎血资源的依赖。该试剂支持 “减少、替代、优化” 的 3R 动物保护原则,不仅解决了资源受限的行业痛点,还能保障长期稳定供应,为内毒素检测领域的可持续发展提供了理想解决方案。
样品中的脂质体与表面活性剂,会通过不同机制干扰内毒素检测,需结合基质特性选择处理方式。脂质体的干扰体现在两方面:一是脂质双分子层会包裹内毒素,使其无法与鲎试剂接触,导致假阴性;二是脂质体的胶体性质会干扰凝胶形成(凝胶法)或光度信号读取(浊度 / 显色法)。针对完整脂质体制剂,可先用生理盐水稀释降低脂质体浓度,减少包裹作用;若需彻底释放内毒素,还可使用 DMSO、乙醇或甲醇等溶剂裂解脂质体,暴露被包裹的内毒素。表面活性剂的干扰则源于其对物质结构的改变:既可能破坏内毒素的聚集体结构,影响其活性;又可能导致鲎试剂中蛋白质变性,抑制反应。对此,可通过内毒素检查用水或生理盐水稀释样品,降低表面活性剂浓度,消除其对结构的破坏作用。这些针对性处理能有效解决脂质体与表面活性剂的干扰,确保内毒素检测结果真实可靠。
内毒素检测方法学验证需覆盖线性、精密度,确保不同批次检测结果稳定。
在进行内毒素检测时,干扰试验又叫增强或抑制试验,主要目的是确证检测内毒素的方法是否受样品干扰。在建立细菌内毒素检查方法中,验证试验前,要去除样品可能含有的内毒素,以确保建立方法的准确可靠。药典规定:①当进行新药的内毒素检查试验前,或无内毒素检查项品种建立内毒素检查法时,需进行干扰试验;②当鲎试剂、供试品的配方、生产工艺改变,或试验环境下发生了任何有可能影响试验结果的变化时,需重新进行干扰试验。生产厂家常发生的一些微小变更,会影响到评估结果,进而影响到供试品对鲎试剂的干扰试验。因此,生产厂家应制定一个重复进行干扰试验的周期,并进行跟踪和记录。
细菌内毒素检查有凝胶法和光度测定法,结果有争议时,除规定外以凝胶限度试验为准。上海疫苗内毒素检测技术服务
重组级联试剂(rCR)无动物源,不含 G 因子,可避免 β- 葡聚糖致内毒素检测假阳性。北京合规性内毒素检测低内毒素回收
湖州申科建立了标准化的低内毒素回收(LER)技术服务流程,全周期支撑内毒素检测优化:7 个自然日内完成客户沟通与 LER 确认,用天然鲎、重组鲎等方法检测并出具报告;60 个自然日开展方法开发,分析 LER 成因(如螯合剂、蛋白质 IP)并研究去掩蔽方案;30 个自然日进行 HTS 验证,完成 3 批 LER 实验与干扰实验,交付稳定试剂盒、操作规程及培训;再协助方法转移与 cGMP 验证。流程每环节均围绕内毒素检测展开,确保企业高效解决 LER 问题,满足法规要求。
北京合规性内毒素检测低内毒素回收