湖州申科生物的产品研发与制造过程遵循 ISO13485 质量管理体系要求开展,并参照 CDE《生物制品质量控制分析方法验证技术审评一般原则》、《中国药典》9101 分析方法验证指导原则及 ICH Q2 (R2)《分析方法验证指导原则》等法规文件,对 SHENTEK 系列宿主细胞残留检测试剂盒实施了全面性能验证,涵盖线性、范围、准确性、定量限、精密度、耐用性、专属性等项目,结果符合法规标准。公司可向用户提供试剂盒的详细验证报告,用户若采用此验证报告,只需开展针对样品的适用性验证(含精密度实验与回收率实验)即可。
检测宿主细胞残留 DNA 时,其扩增曲线需呈正常 “S” 型以保障有效。安徽Human宿主细胞残留DNA检测方案
SHENTEK®CV-1残留DNA检测试剂盒(PCR-荧光探针法),可对各类生物制品的中间品、半成品及成品中CV-1宿主细胞DNA进行定量检测。该试剂盒基于荧光探针法原理,实现对样品中CV-1残留DNA的定量分析,具备检测快速、专一性强、性能稳定可靠的特点,检测限可达到fg级别。试剂盒内配套有CV-1 DNA定量参考品,且与SHENTEK®宿主细胞残留DNA样本前处理试剂盒搭配使用,可准确定量样品中的CV-1残留DNA。整个分析系统通过优化前处理与检测步骤的兼容性,能有效提升对CV-1细胞微量DNA残留的回收率及定量准确度。
安徽Human宿主细胞残留DNA检测方案湖州申科围绕宿主细胞残留 DNA 检测,专注于多种生物分析方法的研发优化,遵照法规与指导原则执行。
宿主细胞残留DNA检测体系(qPCR法)的技术关键点涉及多个维度。1、靶标序列的查找与确定:需参照宿主物种的基因组序列,筛选出物种特异性强、高度重复且呈散在分布的序列,将其作为检测靶标。2、检测体系建立与方法验证:在适宜位点设计引物与探针,选择适配的荧光及淬灭基团。3、基于qPCR法优化体系组分比例,制备用于微量DNA模板检测的MIX,同时对线性范围、专属性等多项指标开展验证。4、检测体系稳定性保障:需完成参考品的准确标定,同时严格控制试剂批间差异,确保检测体系的稳定性。5、适配的残留DNA提取体系:需搭配合适的宿主细胞残留DNA提取体系,以应对不同样品基质的影响,实现微量残留DNA的回收提取与纯化。
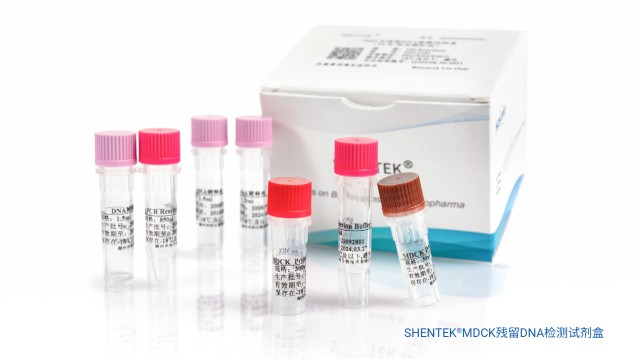
MDCK 细胞作为宿主工程用细胞,其宿主细胞DNA残留会给机体带来安全风险。虽然 MDCK 细胞基质流感疫苗生产中,已采用超滤、核酸酶处理、层析、β- 丙内酯灭活等工艺去除残留 DNA,但疫苗产品里仍可能残留宿主细胞DNA 及其片段。所以,为把控疫苗质量,需对疫苗中的 MDCK 宿主细胞残留 DNA 及其片段开展检测与分析。湖州申科生物推出 MDCK 残留 DNA 检测试剂盒与 MDCK DNA 残留片段分析检测试剂盒,帮助相关疫苗企业对 MDCK 流感疫苗的 HCD 实施全过程监控。SHENTEK® 系列宿主细胞残留 DNA 检测试剂盒,与 SHENTEK-96S 等常用 PCR 设备适配。
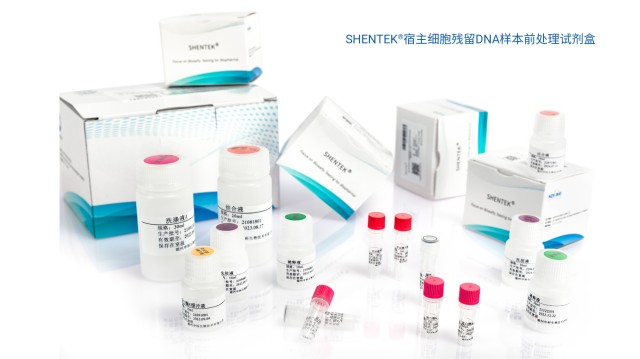
宿主细胞残留DNA样本前处理试剂盒(磁珠法)机装版,适用于生物制品样品前处理,能稳定高效获取样品中的微量宿主细胞DNA。该试剂盒可在多种复杂基质缓冲液(如细胞裂解液、澄清上清、纯化中间品等)中,实现DNA的稳定提取与高回收率。同时,其适配多种基质缓冲溶液,能有效提取纯化微量DNA,可与各类SHENTEK®宿主细胞(CHO、大肠杆菌E.coli、Vero、酵母、NS0、Human、MDCK、Sf9&AcNPV、Hi5&AcNPV、质粒、SV40LTA&EIA等)DNA qPCR检测试剂盒搭配使用。
检测宿主细胞残留 DNA 所获数据,是产品放行的关键依据之一。
安徽Human宿主细胞残留DNA检测方案生物制品宿主细胞残留 DNA 可能存在污染风险,将对产品使用者带来潜在安全性影响。安徽Human宿主细胞残留DNA检测方案
宿主细胞残留DNA(rDNA)的风险研究,主要涵盖传染性、致病性、免疫原性等方面,各国药典均对其残留量设定了严格限度要求:美国食品药品监督管理局(FDA)在指导原则中明确,生物制品的宿主细胞DNA残留限度需≤100pg/剂;针对单克隆抗体等大剂量生物制品,可依据残留DNA来源及给药途径,将限度放宽至10ng/剂。《欧洲药典》通则中,多数生物制品的残留DNA限度规定为不超过10ng/剂。2025版《中国药典》三部则规定,采用细胞基质生产的生物制剂,其DNA残留量不得超过100pg/剂。编辑分享中国药典对宿主细胞残留DNA(rDNA)的限度要求是如何规定的?请给出一段关于宿主细胞残留DNA风险研究的简介。宿主细胞残留DNA的检测方法有哪些?
安徽Human宿主细胞残留DNA检测方案