质控结果是支原体 NAT 检测可靠性的前提,需严格遵循判定标准,且需结合实验室实际条件验证适配的标准阈值。质控样品的判定规则明确:NTC(无模板对照)的 FAM 信号需 2 复孔 Ct≥40 或无明显扩增曲线,VIC 信号需 2 复孔 Ct<35 且呈有效 “S” 型扩增;NCS(阴性对照)判定标准与 NTC 一致;PC(阳性质控)的 FAM 信号需 2 复孔 Ct<35 且呈有效 “S” 型扩增,PCS(阳性对照底物)判定标准与 PC 一致。只有质控结果全部满足要求,才能进一步分析样本结果,若质控不达标,需排查设备、试剂、操作等环节的问题并重新检测。
耐用性验证需确认方法参数微小变动时,支原体检测结果不受影响。上海疫苗产品支原体检测培养法
支原体 NAT 检测中常见三类问题,其成因多与样品基质、操作规范或污染防控相关。1、样品基质干扰,高浓度 DMSO、人血白蛋白等冻存保护剂,高浓度细胞、DNA 碎片等复杂成分,会抑制检测反应或干扰信号读取。2、检测曲线异常,表现为非特异性扩增图谱,多因引物设计不当、反应条件优化不足或试剂交叉污染导致。3、阴性污染,具体体现为无模板对照(NTC)、阴性对照(NCS)出现翘尾现象,主要源于实验室分区不合理、操作流程不规范(如阴阳性样本交叉处理)、耗材污染或环境气溶胶污染,这些问题均会影响结果判定的准确性,需针对性解决。
湖南重组药物支原体检测国产替代培养基、血清等原辅料入库前需做支原体检测,排除外源污染风险。
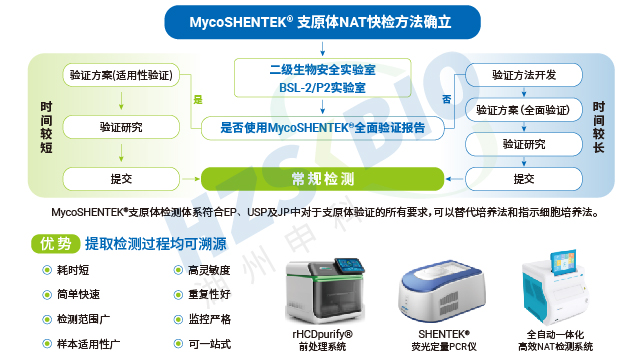
支原体 NAT 检测的特异性验证面临主要挑战:需设计覆盖多种支原体的 PCR 引物,而覆盖范围越广,越可能因支原体与革兰氏阳性菌的系统进化相关性,出现交叉检测现象,影响结果准确性。因此,特异性验证需重点排查非目标微生物的干扰,确保检测结果的专一性。稳健性验证同样关键,需证实检测方法在人为引入的微小参数变化(如反应温度、试剂浓度波动等)下,仍能保持结果稳定,避免因实验条件差异导致的检测偏差。这两项验证直接决定了 NAT 方法在不同实验室、不同操作场景下的适用性,是其获得法规认可的重要前提。
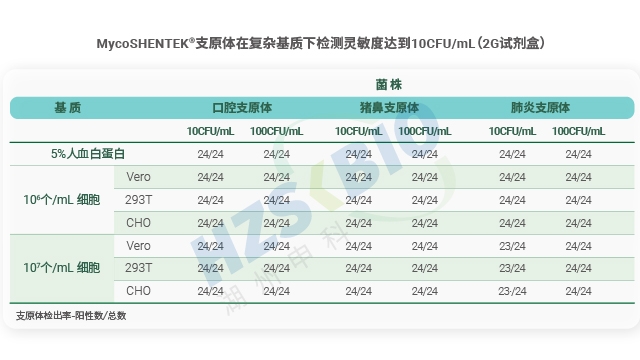
MycoSHENTEK® 支原体 qPCR 检测试剂盒(2G)完全符合 EP 2.6.7 的全部验证要求,其检测灵敏度、特异性、耐用性均按药典标准完成完整性能验证,具备替代培养法和指示细胞培养法的合规资质。该试剂盒针对新型生物制品的检测痛点优化升级,经多种支原体菌株验证,灵敏度稳定达到 10 CFU/mL,满足法规对替代培养法的要求。同时,产品遵循 ISO13485 体系认证和 GMP-like 生产标准,可提供完整的验证报告、质检报告及菌株溯源文件,全程贴合各国药典监管要求,为企业合规检测提供坚实支撑。
复杂样品在进行支原体检测时,可适当稀释样本或增加蛋白酶 K 用量,消除基质干扰。
菌株质量是支原体检测 NAT 方法验证合规的关键,GC/CFU 比(基因组拷贝数与菌落形成单位比值)是关键控制指标。支原体存在聚集特性,单一 CFU 可能对应多个菌体,且 DNA 复制与细胞分裂不同步,部分菌体无法形成菌落但会释放 DNA,导致 GC/CFU 比波动大(研究报道 0.1 CFU 对应 30-500 个基因组拷贝)。若使用高 GC/CFU 比菌株,会高估检测限、导致方法灵敏度不足,因此法规明确要求菌株 GC/CFU 比<10。2024 年 EDQM 36.1 草案规定参考品 GC/CFU 比应小于 10,USP 77 征求意见稿要求表征菌株 GC/CFU 比、建立菌株库 CFU 与核酸拷贝数关系,JP G3-14-170 也对菌株质量提出明确要求,凸显合规菌株选择的重要性。
支原体检测过程中,需严格遵循 “先阴后阳” 操作原则,避免交叉污染。山东疫苗产品支原体检测验证菌株
支原体检测 NAT 法引物设计需平衡覆盖范围与特异性,避免交叉反应。上海疫苗产品支原体检测培养法
支原体 NAT 检测中异常曲线的出现多与操作设置、耗材使用或实验环境相关,需针对性排查解决。首先需确认软件设置正确性,对照试剂盒说明书检查时间、温度、循环数、荧光采集等参数,尤其需注意关闭不含 ROX 试剂的参比荧光功能。其次若扩增曲线无抬升却出现 CT 值,需调整基线范围,将起点设为荧光信号稳定的循环数,终点设为扩增曲线起峰前一个循环数。再者需确保耗材与仪器匹配,避免使用普通 PCR 耗材或与加热模块规格不符的反应管,防止荧光传导不佳或热传导不充分。此外,曲线先上升后下降可能是反应液蒸发导致,上机前需检查八连管管盖无缝隙,避免液面下降干扰结果。
上海疫苗产品支原体检测培养法