支原体 NAT 检测中异常曲线的出现多与操作设置、耗材使用或实验环境相关,需针对性排查解决。首先需确认软件设置正确性,对照试剂盒说明书检查时间、温度、循环数、荧光采集等参数,尤其需注意关闭不含 ROX 试剂的参比荧光功能。其次若扩增曲线无抬升却出现 CT 值,需调整基线范围,将起点设为荧光信号稳定的循环数,终点设为扩增曲线起峰前一个循环数。再者需确保耗材与仪器匹配,避免使用普通 PCR 耗材或与加热模块规格不符的反应管,防止荧光传导不佳或热传导不充分。此外,曲线先上升后下降可能是反应液蒸发导致,上机前需检查八连管管盖无缝隙,避免液面下降干扰结果。
湖州申科支原体检测试剂盒通过FDA DMF备案,支持全球药品申报使用。广东免疫细胞产品支原体检测使用性验证
支原体 NAT 检测的特异性验证面临主要挑战:需设计覆盖多种支原体的 PCR 引物,而覆盖范围越广,越可能因支原体与革兰氏阳性菌的系统进化相关性,出现交叉检测现象,影响结果准确性。因此,特异性验证需重点排查非目标微生物的干扰,确保检测结果的专一性。稳健性验证同样关键,需证实检测方法在人为引入的微小参数变化(如反应温度、试剂浓度波动等)下,仍能保持结果稳定,避免因实验条件差异导致的检测偏差。这两项验证直接决定了 NAT 方法在不同实验室、不同操作场景下的适用性,是其获得法规认可的重要前提。
湖南免疫细胞产品支原体检测快速检测支原体标准菌株对支原体检测试剂盒的选择、使用及验证至关重要。
湖州申科围绕 USP 支原体培养法,提供技术服务,满足生物制剂企业的合规需求。服务内容包括两项:一是支原体培养法样品检测服务,严格遵循 USP 63 标准流程执行,确保检测结果合规有效;二是支原体培养法样品适用性验证服务,针对供试品特性开展定制化验证,保障检测方法与样品的适配性。所有检测与验证完成后,公司将出具标准化报告,报告可直接用于项目申报、审批、工艺优化等场景,为企业产品质量控制提供依据。通过专业的技术服务,帮助企业规避合规风险,确保生物制剂生产全流程的质量与安全性,助力企业高效推进产品研发与上市进程。
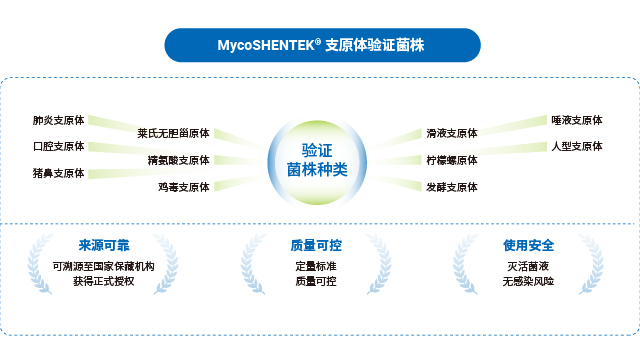
菌株质量是支原体检测 NAT 方法验证合规的关键,GC/CFU 比(基因组拷贝数与菌落形成单位比值)是关键控制指标。支原体存在聚集特性,单一 CFU 可能对应多个菌体,且 DNA 复制与细胞分裂不同步,部分菌体无法形成菌落但会释放 DNA,导致 GC/CFU 比波动大(研究报道 0.1 CFU 对应 30-500 个基因组拷贝)。若使用高 GC/CFU 比菌株,会高估检测限、导致方法灵敏度不足,因此法规明确要求菌株 GC/CFU 比<10。2024 年 EDQM 36.1 草案规定参考品 GC/CFU 比应小于 10,USP 77 征求意见稿要求表征菌株 GC/CFU 比、建立菌株库 CFU 与核酸拷贝数关系,JP G3-14-170 也对菌株质量提出明确要求,凸显合规菌株选择的重要性。
支原体检测中,检测限验证需对每种支原体做3次10倍梯度稀释,获取24个数据满足95%检出率。
支原体 NAT 检测的样本结果需结合 FAM 信号与 VIC 信号的表现综合判定,同时警惕抑制现象对结果的影响。若 FAM 信号 2 复孔有 1 孔以上 Ct<40 且呈有效 “S” 型扩增,VIC 信号 2 复孔 Ct<40 且呈有效 “S” 型扩增,判定为阳性;若 VIC 信号 2 复孔 Ct≥40 或无明显扩增曲线,则为阳性但存在抑制。若 FAM 信号 2 复孔 Ct≥40 或无明显扩增曲线,VIC 信号 2 复孔 Ct<40 且呈有效 “S” 型扩增,判定为阴性;若 VIC 信号同样 Ct≥40 或无明显扩增曲线,则阴阳性无法判断且存在抑制。出现抑制现象需重测,重测仍有抑制时,阳性倾向样本可适当稀释,阴性倾向样本需优化提取条件。
支原体检测NAT法(核酸扩增法)周期只需 3-6 小时,灵敏度高、特异性强,是生物制品快检优先选择方案。上海免疫细胞产品支原体检测验证菌株
疫苗病毒种子批支原体检测需取全量样品,建议进行 10mL 种子液提取,确保无漏检。广东免疫细胞产品支原体检测使用性验证
MycoSHENTEK® 支原体 qPCR 检测试剂盒(2G)完全符合 EP 2.6.7 的全部验证要求,其检测灵敏度、特异性、耐用性均按药典标准完成完整性能验证,具备替代培养法和指示细胞培养法的合规资质。该试剂盒针对新型生物制品的检测痛点优化升级,经多种支原体菌株验证,灵敏度稳定达到 10 CFU/mL,满足法规对替代培养法的要求。同时,产品遵循 ISO13485 体系认证和 GMP-like 生产标准,可提供完整的验证报告、质检报告及菌株溯源文件,全程贴合各国药典监管要求,为企业合规检测提供坚实支撑。
广东免疫细胞产品支原体检测使用性验证